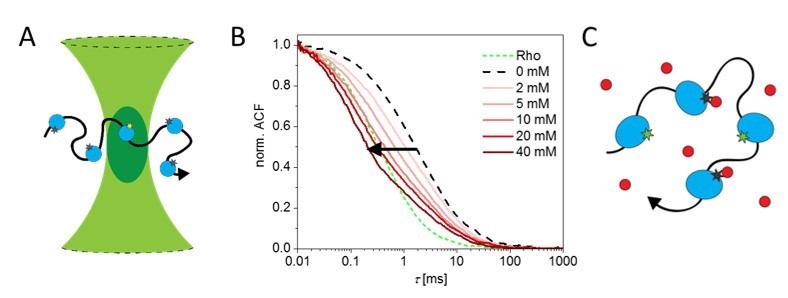
Figure: A) Principle of fluorescence correlation spectroscopy; the transitions of a single enzyme (blue) through a confocal volume (green) is observed due to the fluorescence of the dye (asterisk); B) for the enzyme alkaline phosphatase an apparent decrease in diffusion can be observed with increasing concentrations of its substrate [1]; However, we showed that C) an apparent decrease in transition time can also be caused by the interaction of a quencher (red) with the fluorophore by shifting it into a non-fluorescent dark state [2].
Enzymes
Jan-Philipp Günther, Peer Fischer
Collaborators: Michael Börsch and Günter Majer
Chemical reactions on the surface of microparticles can cause local concentration gradients near the particle. A local gradient of substrate or product molecules can in turn give rise to fluid flows around the particle, and momentum conservation then requires that the particle moves in the opposite direction to these flows. Energy is consumed, which powers the self-propelled particles. The particles are therefore a model system for “active matter” and are also known as chemical motors, which can autonomously swim and self-organize [3,4]. If these or related mechanisms can also translate to the molecular scale is, however, not yet fully understood. The question whether the reaction of enzymes can cause their self-propulsion has potentially important implications for biochemistry. Similarly, the question whether these systems can show chemotaxis has far reaching implications as they would suggest a radically new mechanism how one may realize drug delivery, e.g. by means of active self-propelled carriers that can steer themselves to the location they are needed [5].
Fluorescence correlations spectroscopy (FCS) can be used to determine the diffusion of fluorescently labeled biomolecules. Together with our collaborator Michael Boersch (Jena) we have performed precise measurements of enzyme diffusion to examine if enzymes can self-propel. We used an advanced optical setup to elucidate how the fluorescence dye interacts with its environment and the enzyme it is coupled to. We also introduced in situ fluorescence lifetime analysis via time-correlated photon counting (TCPC), and we measured the anisotropy of the fluorescence signal. The former provides information regarding the quantum states of the dye and the latter can provide information about its binding to the enzyme. Additionally we considered serval biochemical factors that could affect the FCS signals.
The highest diffusion enhancement for a catalytically enzyme has been reported for the enzyme alkaline phosphatase [1]. We find that a significant decrease in the transition time through the confocal volume of the FCS setup can indeed be observed occurs (Figure panel B), but in situ life-time measurements also show that quenching of the fluorophore by the substrate of alkaline phosphate occurs. Quenching effects add additional fluctuations to the fluorescence signal, which can thus lead to the erroneous conclusion that the enzyme shows enhanced diffusion. We find no diffusion enhancement by alkaline phosphatase related to its activity – this enzyme does not seem swim when it is catalytically active [2].
The insight that enzymes do not exhibit activity-enhanced diffusion provides size limits for active matter and self-propelled swimmers. Our ongoing studies using Nuclear Magnetic Resonance lend further support to our findings [6]. It is thus an open question how small a swimmer can be for it to efficiently self-propel. We are currently investigating active nanoparticles to address this question as well as how one can design autonomous nanomotors that can chemotax.
Literature:
[1] C. Riedel, R. Gabizon, C.A.M. Wilson, K. Hamadani, L. Tsekouras, S. Marqusee, S. Presse, C. Bustamante, Nature 517, 227, (2015).
[2] J.-P. Günther, M. Börsch, P. Fischer, Acc. Chem. Res. 51, 1911−1920, (2018).
[3] D. P. Singh, U. Choudhury, P. Fischer, A. G. Mark, Advanced Materials 29: 1701328, (2017).
[4] D.P. Singh, W.E. Uspal, M.N. Popescu, L.G. Wilson, P. Fischer, Adv. Func. Mat., 28: 1706660, (2018).
[5] M. N. Popescu, W. E. Uspal, C. Bechinger, P. Fischer, Nano Letters, 18:5345, 2018.
[6] J.-P. Günther, G. Majer, P. Fischer, https://arxiv.org/abs/1812.08748 (to appear in J. Chem. Phys. 2019)